- CATALYSE - Généralités
- CATALYSE - GénéralitésLe terme catalyse a été forgé par Berzelius en 1835 pour désigner l’ensemble des effets chimiques produits par les catalyseurs. On appelle catalyseur «toute substance qui altère la vitesse d’une réaction chimique sans apparaître dans les produits finaux» (Ostwald, 1902). La catalyse est donc une branche de la cinétique chimique qui, de façon plus générale, étudie l’influence de tous les facteurs physiques ou chimiques déterminant les vitesses de réaction.La notion de catalyseur est intuitive et elle est passée dans le langage courant pour désigner, par exemple, une personne physique ou morale provoquant par son action ou son exemple une importante transformation politique ou sociale, dont elle n’est pas le moteur.La définition du catalyseur s’est précisée peu à peu et s’est progressivement distinguée de celle assez voisine de l’initiateur .Un initiateur est une substance qui produit aussi l’accélération de certains processus chimiques, mais, ce faisant, il est nécessairement détruit. Les peroxydes organiques sont, par exemple, des initiateurs de la polymérisation du styrolène liquide en polystyrènes solides (matières plastiques).Il en est de même des inhibiteurs , improprement nommés catalyseurs négatifs, ou anticatalyseurs, qui ralentissent fortement ou même suppriment certaines réactions. Des antioxygènes tels que les alcoylphénols inhibent l’oxydation du caoutchouc par l’oxygène de l’air.Les peroxydes initiateurs et les alcoylphénols sont consommés et chimiquement dénaturés au cours de leur action. Au contraire, un catalyseur reste identique à lui-même au cours de la catalyse et peut ainsi, en principe, être indéfiniment réutilisé. Par exemple, le platine ou le palladium divisés catalysent la combinaison des gaz hydrogène et oxygène en eau (Davy, 1817). Moyennant certaines précautions, ces catalyseurs peuvent provoquer la combinaison de quantités aussi grandes que l’on veut d’hydrogène et d’oxygène.En pratique néanmoins, par suite de réactions parasites, le catalyseur a une durée de vie limitée; son action s’affaiblit au cours du temps et s’interrompt complètement au bout de quelques heures ou de quelques années. Cette désactivation n’est pas intrinsèquement liée au mécanisme de la catalyse, mais à des effets physiques (haute température) ou chimiques (présence d’impuretés) parfois réversibles, la réactivation étant alors possible par des procédés simples. La longévité d’un catalyseur peut être mesurée par la quantité de matière qu’il a transformée par unité de masse avant sa mort: ce rapport théoriquement infini est souvent très élevé (102 à 106). S’il est très faible (quelques unités), on peut se demander s’il y a vraiment catalyse.On distingue la catalyse homogène et la catalyse hétérogène ; dans le premier cas, le catalyseur est dissous dans le milieu (gaz ou liquide) où se produit la réaction; dans le second, le catalyseur est un solide au contact de ce milieu, d’où le nom de catalyse de contact. La séparation du catalyseur des produits de la réaction est alors grandement simplifiée.L’importance pratique de la catalyse dans l’industrie chimique est considérable. Presque tous les produits de grande consommation sont passés par une catalyse à un ou plusieurs stades de leur élaboration: essences et huiles de voiture (cracking, reforming, hydrofining), détergents (alcoylation), caoutchoucs, matières plastiques et fibres synthétiques (synthèse des monomères et polymérisation) et même notre pain quotidien, qui, produit direct d’une catalyse enzymatique, est le produit indirect des engrais azotés de synthèse (catalyse de l’ammoniac).1. Thermodynamique et catalyseL’effet du catalyseur ne peut être que cinétique; par définition, il se retrouve en effet intact en fin de réaction: sa présence ne peut donc modifier les grandeurs thermodynamiques caractéristiques (variations d’enthalpie H 0, d’enthalpie libre G 0) et la constante d’équilibre liée à ces grandeurs. Les conséquences de cette remarque sont très importantes. En premier lieu, il ne faut pas espérer obtenir, en présence d’un catalyseur, une réaction thermodynamiquement impossible dans les conditions de l’expérience ( G 0 positif et grand). Ainsi, dans les conditions habituelles, la combinaison de l’azote et de l’oxygène de l’air en oxydes d’azote (N2O, NO, 2) ne peut se faire, quel que soit le catalyseur envisagé, circonstance heureuse pour notre espèce. En revanche, la combinaison de l’hydrogène et de l’oxygène est possible, mais sa vitesse est nulle dans les mêmes circonstances, sauf en présence d’un catalyseur adéquat. Celle de l’azote et de l’hydrogène en ammoniac l’est aussi, mais, jusqu’à ce jour, aucun catalyseur n’a été trouvé capable de lui donner une vitesse notable au voisinage de la température ambiante. Cet important problème n’est pas résolu, mais il n’est pas insoluble.En deuxième lieu, puisque le catalyseur ne peut déplacer la position finale d’un équilibre chimique, mais qu’il diminue fortement le temps nécessaire pour l’atteindre, c’est qu’il accélère dans le même rapport les vitesses des deux réactions inverses dont l’égalité est la condition de l’équilibre. Ainsi, le même catalyseur doit accélérer la synthèse et la décomposition de l’ammoniac en ses éléments, le fer par exemple. On peut alors trouver le catalyseur d’une réaction donnée en recherchant le catalyseur de la réaction inverse, pour les couples hydrogénation-déshydrogénation, hydratation-déshydratation. Pour la même raison, l’influence de la quantité de catalyseur (plus précisément de sa concentration) sur les deux réactions inverses doit s’exprimer par la même loi, et les formules cinétiques exprimant leurs vitesses doivent contenir les produits de concentrations de la loi d’action de masse.Soit la réaction:
 la condition thermodynamique de l’équilibre est:
la condition thermodynamique de l’équilibre est: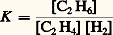 et la condition cinétique: v1 = v2.Pour que ces deux conditions soient identiques, on peut avoir:
et la condition cinétique: v1 = v2.Pour que ces deux conditions soient identiques, on peut avoir: avec K = k 1/k 2, f (K ) fonction de la concentration en catalyseur K et g () fonction des concentrations des corps en réaction. Les fonctions f (K ) et g (K ) s’éliminent ainsi dans l’égalité cinétique. La forme des fonctions f et g , et la valeur absolue de k 1 et k 2 varient en général avec le catalyseur utilisé, et ces facteurs dépendent de la température. Ainsi, pour certains catalyseurs et dans un domaine limité des variables, on aurait:
avec K = k 1/k 2, f (K ) fonction de la concentration en catalyseur K et g () fonction des concentrations des corps en réaction. Les fonctions f (K ) et g (K ) s’éliminent ainsi dans l’égalité cinétique. La forme des fonctions f et g , et la valeur absolue de k 1 et k 2 varient en général avec le catalyseur utilisé, et ces facteurs dépendent de la température. Ainsi, pour certains catalyseurs et dans un domaine limité des variables, on aurait: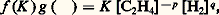 Dans tous les cas très simples qui précèdent, l’évolution du système chimique initial est pratiquement univoque. Mais, dans beaucoup d’autres cas, le système peut évoluer dans plusieurs directions thermodynamiquement permises; cette situation est particulièrement fréquente avec les composés du carbone (chimie organique). Un catalyseur approprié peut alors accélérer de préférence une seule de ces évolutions, et cette sélectivité est une propriété souvent plus précieuse que l’effet accélérateur global, qui mesure son activité. Par exemple, un mélange gazeux d’oxyde de carbone et d’hydrogène peut, suivant le catalyseur choisi, être converti en méthane (nickel), méthanol (oxyde de zinc), ou n -alcanes (synthèse Fischer-Tropsch, catalyseur au cobalt).Cela ne contredit nullement la thermodynamique, et un catalyseur très sélectif permet l’étude d’équilibres partiels qui seraient, en son absence, noyés dans un système très compliqué de réactions parallèles ou successives. On conçoit aisément l’importance pratique de cette sélectivité dans les synthèses industrielles.2. Mécanisme général de l’action des catalyseursLe mot catalyse a longtemps servi à cacher l’ignorance complète où se trouvaient les chimistes devant certains phénomènes inexplicables. L’action des catalyseurs entre en fait dans le cadre général de l’étude des mécanismes réactionnels, c’est-à-dire de la recherche des étapes successives par lesquelles les molécules des corps mis en réaction se transforment pour donner les produits finaux.Ces étapes comportent chacune la rupture ou la formation d’un petit nombre de liaisons chimiques entre atomes, et mettent souvent en jeu des intermédiaires très instables et rarement isolables, comme les radicaux libres ou les ions.La présence du catalyseur modifie le mécanisme réactionnel et ouvre à la transformation une voie souvent plus complexe mais finalement plus rapide, comportant des étapes à faible énergie d’activation. Cela n’est possible que dans la mesure où le catalyseur est lui-même un réactif pour une ou plusieurs de ces étapes, et un produit pour une ou plusieurs autres: il est donc constamment consommé et régénéré au cours de la catalyse, et il ne reste intact qu’en apparence. Cela est illustré schématiquement dans la figure 1.Généralement, le catalyseur réagit avec les molécules de réactif les plus inertes (stables) et les fait entrer dans le cycle réactionnel; c’est ce que l’on exprime en disant que le catalyseur active ces molécules. Ainsi, un catalyseur d’oxydation «active» la molécule d’oxygène, un catalyseur d’hydrogénation celle d’hydrogène. Cette activation comporte ou non une dissociation de la molécule activée. Les atomes du catalyseur sur lesquels portent ces réactions sont appelés centres actifs .Les étapes ultérieures libèrent les centres actifs, qui deviennent ainsi à nouveau disponibles pour recommencer le cycle. Par exemple, la molécule à oxyder ou à hydrogéner réagit avec l’oxygène ou l’hydrogène lié au catalyseur. Elle peut elle-même avoir été au préalable activée par un processus semblable. La nature et la réactivité des intermédiaires résultant de la réaction des centres actifs avec les molécules initiales déterminent l’activité et la sélectivité du catalyseur.Il est relativement facile d’identifier les centres actifs et les espèces intermédiaires dans la catalyse homogène, par les méthodes habituelles de la chimie. La tâche est beaucoup plus difficile en catalyse hétérogène, car ces centres actifs font partie de la surface d’un solide, auquel ils restent liés, mais l’existence de ces centres est bien connue, et elle explique notamment les phénomènes d’adsorption chimique. Toutefois, le rapprochement entre catalyses homogène et hétérogène, et l’emploi des méthodes physico-chimiques les plus modernes permettent d’affirmer que les différences entre ces deux classes de phénomènes ne sont pas aussi grandes que l’on avait pensé.3. Catalyse homogènePar définition, le catalyseur est dissous dans le gaz, le liquide ou le solide dans lequel se produit la réaction catalysée. C’est donc un corps à l’état moléculaire, que l’on connaît bien puisque l’opérateur lui-même l’a placé dans le mélange.Dans un gaz, le catalyseur est forcément une molécule assez simple, ce qui permet de supposer qu’on expliquera facilement le mécanisme: en effet, une molécule très complexe et très lourde ne peut passer facilement à l’état gazeux. En solution, le catalyseur peut être beaucoup plus complexe, et la nature du solvant joue un rôle déterminant. Un cas extrême est celui de la catalyse par les enzymes des principales réactions chimiques accompagnant le fonctionnement des organismes vivants (digestion, respiration). L’activité et la sélectivité de ces catalyseurs naturels dépassent de très loin celles de tous les catalyseurs connus. Leur dimension est telle que l’on se trouve à la limite entre catalyse homogène et hétérogène (plusieurs milliers de fois celle des molécules qu’ils activent).MécanismeOn peut assez souvent, dans les cas simples, séparer la réaction catalytique en deux parties, la première consommant le catalyseur et la deuxième le régénérant. C’est même là un moyen général de «fabriquer» des réactions catalytiques.Ainsi, vers 600 0C, la vapeur d’iode s’empare de l’hydrogène des alcanes pour former de l’acide iodhydrique et des alcènes ou des hydrocarbures dérivés du benzène, de rapport H/C plus faible. D’autre part, l’oxygène attaque l’acide iodhydrique en lui arrachant son hydrogène, pour former de l’eau et de l’iode. On conçoit que, dans les conditions choisies pour que ces deux réactions aient lieu à la même vitesse, l’iode soit un catalyseur de déshydrogénation des alcanes par l’oxygène, puisque:
Dans tous les cas très simples qui précèdent, l’évolution du système chimique initial est pratiquement univoque. Mais, dans beaucoup d’autres cas, le système peut évoluer dans plusieurs directions thermodynamiquement permises; cette situation est particulièrement fréquente avec les composés du carbone (chimie organique). Un catalyseur approprié peut alors accélérer de préférence une seule de ces évolutions, et cette sélectivité est une propriété souvent plus précieuse que l’effet accélérateur global, qui mesure son activité. Par exemple, un mélange gazeux d’oxyde de carbone et d’hydrogène peut, suivant le catalyseur choisi, être converti en méthane (nickel), méthanol (oxyde de zinc), ou n -alcanes (synthèse Fischer-Tropsch, catalyseur au cobalt).Cela ne contredit nullement la thermodynamique, et un catalyseur très sélectif permet l’étude d’équilibres partiels qui seraient, en son absence, noyés dans un système très compliqué de réactions parallèles ou successives. On conçoit aisément l’importance pratique de cette sélectivité dans les synthèses industrielles.2. Mécanisme général de l’action des catalyseursLe mot catalyse a longtemps servi à cacher l’ignorance complète où se trouvaient les chimistes devant certains phénomènes inexplicables. L’action des catalyseurs entre en fait dans le cadre général de l’étude des mécanismes réactionnels, c’est-à-dire de la recherche des étapes successives par lesquelles les molécules des corps mis en réaction se transforment pour donner les produits finaux.Ces étapes comportent chacune la rupture ou la formation d’un petit nombre de liaisons chimiques entre atomes, et mettent souvent en jeu des intermédiaires très instables et rarement isolables, comme les radicaux libres ou les ions.La présence du catalyseur modifie le mécanisme réactionnel et ouvre à la transformation une voie souvent plus complexe mais finalement plus rapide, comportant des étapes à faible énergie d’activation. Cela n’est possible que dans la mesure où le catalyseur est lui-même un réactif pour une ou plusieurs de ces étapes, et un produit pour une ou plusieurs autres: il est donc constamment consommé et régénéré au cours de la catalyse, et il ne reste intact qu’en apparence. Cela est illustré schématiquement dans la figure 1.Généralement, le catalyseur réagit avec les molécules de réactif les plus inertes (stables) et les fait entrer dans le cycle réactionnel; c’est ce que l’on exprime en disant que le catalyseur active ces molécules. Ainsi, un catalyseur d’oxydation «active» la molécule d’oxygène, un catalyseur d’hydrogénation celle d’hydrogène. Cette activation comporte ou non une dissociation de la molécule activée. Les atomes du catalyseur sur lesquels portent ces réactions sont appelés centres actifs .Les étapes ultérieures libèrent les centres actifs, qui deviennent ainsi à nouveau disponibles pour recommencer le cycle. Par exemple, la molécule à oxyder ou à hydrogéner réagit avec l’oxygène ou l’hydrogène lié au catalyseur. Elle peut elle-même avoir été au préalable activée par un processus semblable. La nature et la réactivité des intermédiaires résultant de la réaction des centres actifs avec les molécules initiales déterminent l’activité et la sélectivité du catalyseur.Il est relativement facile d’identifier les centres actifs et les espèces intermédiaires dans la catalyse homogène, par les méthodes habituelles de la chimie. La tâche est beaucoup plus difficile en catalyse hétérogène, car ces centres actifs font partie de la surface d’un solide, auquel ils restent liés, mais l’existence de ces centres est bien connue, et elle explique notamment les phénomènes d’adsorption chimique. Toutefois, le rapprochement entre catalyses homogène et hétérogène, et l’emploi des méthodes physico-chimiques les plus modernes permettent d’affirmer que les différences entre ces deux classes de phénomènes ne sont pas aussi grandes que l’on avait pensé.3. Catalyse homogènePar définition, le catalyseur est dissous dans le gaz, le liquide ou le solide dans lequel se produit la réaction catalysée. C’est donc un corps à l’état moléculaire, que l’on connaît bien puisque l’opérateur lui-même l’a placé dans le mélange.Dans un gaz, le catalyseur est forcément une molécule assez simple, ce qui permet de supposer qu’on expliquera facilement le mécanisme: en effet, une molécule très complexe et très lourde ne peut passer facilement à l’état gazeux. En solution, le catalyseur peut être beaucoup plus complexe, et la nature du solvant joue un rôle déterminant. Un cas extrême est celui de la catalyse par les enzymes des principales réactions chimiques accompagnant le fonctionnement des organismes vivants (digestion, respiration). L’activité et la sélectivité de ces catalyseurs naturels dépassent de très loin celles de tous les catalyseurs connus. Leur dimension est telle que l’on se trouve à la limite entre catalyse homogène et hétérogène (plusieurs milliers de fois celle des molécules qu’ils activent).MécanismeOn peut assez souvent, dans les cas simples, séparer la réaction catalytique en deux parties, la première consommant le catalyseur et la deuxième le régénérant. C’est même là un moyen général de «fabriquer» des réactions catalytiques.Ainsi, vers 600 0C, la vapeur d’iode s’empare de l’hydrogène des alcanes pour former de l’acide iodhydrique et des alcènes ou des hydrocarbures dérivés du benzène, de rapport H/C plus faible. D’autre part, l’oxygène attaque l’acide iodhydrique en lui arrachant son hydrogène, pour former de l’eau et de l’iode. On conçoit que, dans les conditions choisies pour que ces deux réactions aient lieu à la même vitesse, l’iode soit un catalyseur de déshydrogénation des alcanes par l’oxygène, puisque: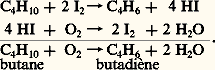 Le bilan de l’opération ne fait plus apparaître l’iode, conformément aux principes de la catalyse. Remarquons qu’en l’absence de catalyseur le butane donne dans les mêmes conditions un mélange complexe de produits de craquage et de combustion inutilisable: la sélectivité est fortement améliorée par l’iode, lorsque le produit désiré est le butadiène, matière première importante des caoutchoucs synthétiques.L’une des plus anciennes fabrications de l’industrie chimique, celle de l’acide sulfurique par le procédé des chambres de plomb, illustre la catalyse d’oxydation du gaz sulfureux par l’air en présence d’oxyde azotique (Clément et Desormes, 1806), schématisée de la même façon:
Le bilan de l’opération ne fait plus apparaître l’iode, conformément aux principes de la catalyse. Remarquons qu’en l’absence de catalyseur le butane donne dans les mêmes conditions un mélange complexe de produits de craquage et de combustion inutilisable: la sélectivité est fortement améliorée par l’iode, lorsque le produit désiré est le butadiène, matière première importante des caoutchoucs synthétiques.L’une des plus anciennes fabrications de l’industrie chimique, celle de l’acide sulfurique par le procédé des chambres de plomb, illustre la catalyse d’oxydation du gaz sulfureux par l’air en présence d’oxyde azotique (Clément et Desormes, 1806), schématisée de la même façon: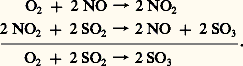 Les deux réactions mises en jeu sont connues et rapides dès la température ambiante, alors que l’oxydation directe du gaz sulfureux par l’air est extrêmement lente à la température des chambres, voisine de 60 0C.Le problème technique le plus sérieux qui se pose dans ces catalyses est la récupération du catalyseur et la ré-injection dans le réacteur, car toute perte doit être compensée par l’addition de catalyseur frais, ce qui grève le prix de revient du produit. Dans les exemples précédents, l’iode peut être séparé de l’hydrocarbure par lavage à l’eau alcalinisée, tandis que l’oxyde azotique est retenu par lavage à l’acide sulfurique de l’air en excès sortant des chambre de plomb (tour de Gay-Lussac)... Cette recirculation du catalyseur complique beaucoup la technologie de la réaction catalysée.Au point de vue scientifique, les schémas précédents ne sont qu’une ébauche très sommaire des mécanismes réactionnels, car chacune des deux réactions écrites comporte en fait plusieurs étapes.Les acides et les basesLes acides et les bases sont les plus simples des catalyseurs de la phase liquide, et les plus anciennement connus.L’acide sulfurique concentré absorbe les alcènes (éthylène, propylène, butylènes) qui, par addition d’eau, sont convertis en alcools: (éthanol, propanol, butanols). La théorie ancienne admettait la formation d’un ester sulfurique, puis son hydrolyse; mais on préfère penser que le vrai catalyseur est le proton H+, car la vitesse de la réaction est directement liée à la concentration de ces ions. Cet exemple illustre bien la réversibilité de l’action catalytique, car, si l’acide sulfurique catalyse à basse température (en dessous de 100 0C) l’addition d’eau sur l’éthylène, il catalyse aussi vers 200 0C la déshydratation de l’éthanol. Ces deux réactions n’ont pas lieu sans catalyseur. Du fait qu’ils agissent comme simples fournisseurs de protons, les acides sont des catalyseurs peu spécifiques, simplement caractérisés par leur «force», qui est la tendance à donner H. Il est d’ailleurs intéressant de rappeler que, historiquement, l’action catalytique des acides sur l’hydrolyse du sucre a été utilisée pour mesurer leur force, à concentrations égales dans l’eau (Wilhelmy, 1850). Puis on a dressé par des moyens autres une échelle de force dite de Hammett dont quelques valeurs sont données ci-dessous:
Les deux réactions mises en jeu sont connues et rapides dès la température ambiante, alors que l’oxydation directe du gaz sulfureux par l’air est extrêmement lente à la température des chambres, voisine de 60 0C.Le problème technique le plus sérieux qui se pose dans ces catalyses est la récupération du catalyseur et la ré-injection dans le réacteur, car toute perte doit être compensée par l’addition de catalyseur frais, ce qui grève le prix de revient du produit. Dans les exemples précédents, l’iode peut être séparé de l’hydrocarbure par lavage à l’eau alcalinisée, tandis que l’oxyde azotique est retenu par lavage à l’acide sulfurique de l’air en excès sortant des chambre de plomb (tour de Gay-Lussac)... Cette recirculation du catalyseur complique beaucoup la technologie de la réaction catalysée.Au point de vue scientifique, les schémas précédents ne sont qu’une ébauche très sommaire des mécanismes réactionnels, car chacune des deux réactions écrites comporte en fait plusieurs étapes.Les acides et les basesLes acides et les bases sont les plus simples des catalyseurs de la phase liquide, et les plus anciennement connus.L’acide sulfurique concentré absorbe les alcènes (éthylène, propylène, butylènes) qui, par addition d’eau, sont convertis en alcools: (éthanol, propanol, butanols). La théorie ancienne admettait la formation d’un ester sulfurique, puis son hydrolyse; mais on préfère penser que le vrai catalyseur est le proton H+, car la vitesse de la réaction est directement liée à la concentration de ces ions. Cet exemple illustre bien la réversibilité de l’action catalytique, car, si l’acide sulfurique catalyse à basse température (en dessous de 100 0C) l’addition d’eau sur l’éthylène, il catalyse aussi vers 200 0C la déshydratation de l’éthanol. Ces deux réactions n’ont pas lieu sans catalyseur. Du fait qu’ils agissent comme simples fournisseurs de protons, les acides sont des catalyseurs peu spécifiques, simplement caractérisés par leur «force», qui est la tendance à donner H. Il est d’ailleurs intéressant de rappeler que, historiquement, l’action catalytique des acides sur l’hydrolyse du sucre a été utilisée pour mesurer leur force, à concentrations égales dans l’eau (Wilhelmy, 1850). Puis on a dressé par des moyens autres une échelle de force dite de Hammett dont quelques valeurs sont données ci-dessous: Cette échelle logarithmique représente un pH généralisé et signifie, par exemple, que l’acide chlorosulfonique HS3Cl pur (face=F0019 漣 12,8) est à peu près cent fois plus fort que l’acide sulfurique H2S4 pur (face=F0019 漣 11).Les catalyseurs dits de Friedel et Crafts (1877) sont des halogénures de métaux. Les mélanges formés par des couples tels que le chlorure d’aluminium et l’acide chlorhydrique (AlCl3 + HCl) ou le fluorure de bore et l’acide fluorhydrique (BF3 + HF) sont des super acides. Ainsi, pour le fluorure de bore en solution à 7 p. 100 dans l’acide fluorhydrique liquide, elle atteint 漣 16,8, soit environ un million de fois celle de l’acide sulfurique pur. Dans de tels milieux, éventuellement additionnés d’un solvant, on a pu identifier récemment (1965), par leurs spectres de résonance magnétique nucléaire, un grand nombre d’ions carbonium stabilisés par une basse température (de 漣 40 à 漣 80 0C), confirmant ainsi l’existence de ces intermédiaires instables postulés depuis longtemps par Whitmore (1930) dans les catalyses acides. Ces ions positifs sont formés par l’action des acides forts sur un grand nombre de corps organiques: alcènes, alcools, chlorures et bromures d’alcoyle. Leurs transformations expliquent un grand nombre de réactions importantes (isomérisation, polymérisation, craquage, alcoylation).La plus ancienne réaction de catalyse par les bases est la saponification des matières grasses, c’est-à-dire la manufacture du savon. Le vrai catalyseur est l’ion hydroxyle OH-(soude ou potasse dans l’eau), qui se combine à la fonction ester en un intermédiaire d’où s’élimine un ion alcoxyle OR-.La force des bases – qui sont dans le plus grand nombre de cas formées d’ions négatifs hydroxyle OH-, alcoxyle OR-, amide NH2-, carbanion R-, associés à un ion positif alcalin Na+, K+ ou Li+ – est mesurée comme celle des acides dans une échelle logarithmique, et dépend considérablement du milieu (solvant). On a trouvé des solvants qui permettent de former des systèmes basiques très puissants: diméthylsulfoxyde, diméthylformamide, hexaméthylphosphoramide. Ainsi, à concentration égale (0,025 molaire), le méthylate de sodium dissous dans un mélange diméthyl-sulfoxyde-méthanol (95/5) est dix millions de fois plus fort que dissous dans le méthanol pur. Dans de tels milieux, un grand nombre de corps organiques sont transformés en ions négatifs, dont les transformations expliquent la catalyse basique. Ainsi, la polymérisation du butadiène en un polybutadiène de très grande longueur (caoutchouc Buna) est catalysée par les organosodiques RNa, R étant un radical alcoyle.Les acides et les bases sont des catalyseurs simples qui accélèrent sans discrimination toutes les réactions passant par l’intermédiaire ionique (ion positif ou négatif) formé lors de leur première attaque des réactifs. Mais leur sélectivité est souvent mauvaise: par exemple, la réaction du benzène avec l’éthylène donnera, à côté de l’éthylbenzène cherché, des di-, tri- et polyéthylbenzènes. La polymérisation du butadiène donne un mauvais caoutchouc, mélange d’isomères appelés polybuta-1,2-diènes et polybuta-1,4-diènes, cis et trans , les unités successives de monomère pouvant s’accrocher à la chaîne en croissance de plusieurs façons différentes.Catalyseurs complexesOn a découvert une classe importante de catalyseurs beaucoup plus sélectifs, appartenant à la famille des complexes solubles de certains métaux, notamment des métaux de transition , qui constituent à certains égards un premier pas vers les enzymes. Ces complexes sont constitués par un atome métallique central entouré d’un certain nombre (2, 3, 4, 5, 6) de molécules ou d’ions qui lui sont attachés par des liaisons chimiques, l’édifice pouvant être au total chargé ou neutre.Ces édifices ont une géométrie simple (fig. 2): tétraèdre, pyramide, octaèdre, carré, pour 4, 5, 6 ou 8 ligands , si l’on appelle ainsi les molécules ou ions liés à l’atome central. Les ligandes sont très variées: ions négatifs tels que Cl-, OH-, C-; molécules telles que H2O, NH3; mais les plus intéressantes pour la catalyse sont par exemple l’oxyde de carbone CO, l’éthylène C2H4, l’acétylène C2H2, l’hydrogène H, les radicaux alcoyles R.En effet, ces corps prennent, dans le complexe, une réactivité particulière: ils sont activés, au sens défini plus haut, et l’atome métallique central constitue un centre actif pour la catalyse.Leur introduction dans le complexe, qui est l’acte fondamental de l’activation, peut se faire par échange de ligandes (substitution), ou par augmentation du nombre de ligandes (addition), processus souvent réversibles. Ainsi, l’activation de la molécule d’hydrogène H2, à froid, est obtenue avec certains complexes de l’argent: [Ag(NR3)2]+ ou du cobalt: [Co(CN)5]3-, en solution aqueuse, avec dissociation de H2 en 2 H, réaction qui ne se produit, en phase gazeuse, qu’à des températures supérieures à 2 000 0C.
Cette échelle logarithmique représente un pH généralisé et signifie, par exemple, que l’acide chlorosulfonique HS3Cl pur (face=F0019 漣 12,8) est à peu près cent fois plus fort que l’acide sulfurique H2S4 pur (face=F0019 漣 11).Les catalyseurs dits de Friedel et Crafts (1877) sont des halogénures de métaux. Les mélanges formés par des couples tels que le chlorure d’aluminium et l’acide chlorhydrique (AlCl3 + HCl) ou le fluorure de bore et l’acide fluorhydrique (BF3 + HF) sont des super acides. Ainsi, pour le fluorure de bore en solution à 7 p. 100 dans l’acide fluorhydrique liquide, elle atteint 漣 16,8, soit environ un million de fois celle de l’acide sulfurique pur. Dans de tels milieux, éventuellement additionnés d’un solvant, on a pu identifier récemment (1965), par leurs spectres de résonance magnétique nucléaire, un grand nombre d’ions carbonium stabilisés par une basse température (de 漣 40 à 漣 80 0C), confirmant ainsi l’existence de ces intermédiaires instables postulés depuis longtemps par Whitmore (1930) dans les catalyses acides. Ces ions positifs sont formés par l’action des acides forts sur un grand nombre de corps organiques: alcènes, alcools, chlorures et bromures d’alcoyle. Leurs transformations expliquent un grand nombre de réactions importantes (isomérisation, polymérisation, craquage, alcoylation).La plus ancienne réaction de catalyse par les bases est la saponification des matières grasses, c’est-à-dire la manufacture du savon. Le vrai catalyseur est l’ion hydroxyle OH-(soude ou potasse dans l’eau), qui se combine à la fonction ester en un intermédiaire d’où s’élimine un ion alcoxyle OR-.La force des bases – qui sont dans le plus grand nombre de cas formées d’ions négatifs hydroxyle OH-, alcoxyle OR-, amide NH2-, carbanion R-, associés à un ion positif alcalin Na+, K+ ou Li+ – est mesurée comme celle des acides dans une échelle logarithmique, et dépend considérablement du milieu (solvant). On a trouvé des solvants qui permettent de former des systèmes basiques très puissants: diméthylsulfoxyde, diméthylformamide, hexaméthylphosphoramide. Ainsi, à concentration égale (0,025 molaire), le méthylate de sodium dissous dans un mélange diméthyl-sulfoxyde-méthanol (95/5) est dix millions de fois plus fort que dissous dans le méthanol pur. Dans de tels milieux, un grand nombre de corps organiques sont transformés en ions négatifs, dont les transformations expliquent la catalyse basique. Ainsi, la polymérisation du butadiène en un polybutadiène de très grande longueur (caoutchouc Buna) est catalysée par les organosodiques RNa, R étant un radical alcoyle.Les acides et les bases sont des catalyseurs simples qui accélèrent sans discrimination toutes les réactions passant par l’intermédiaire ionique (ion positif ou négatif) formé lors de leur première attaque des réactifs. Mais leur sélectivité est souvent mauvaise: par exemple, la réaction du benzène avec l’éthylène donnera, à côté de l’éthylbenzène cherché, des di-, tri- et polyéthylbenzènes. La polymérisation du butadiène donne un mauvais caoutchouc, mélange d’isomères appelés polybuta-1,2-diènes et polybuta-1,4-diènes, cis et trans , les unités successives de monomère pouvant s’accrocher à la chaîne en croissance de plusieurs façons différentes.Catalyseurs complexesOn a découvert une classe importante de catalyseurs beaucoup plus sélectifs, appartenant à la famille des complexes solubles de certains métaux, notamment des métaux de transition , qui constituent à certains égards un premier pas vers les enzymes. Ces complexes sont constitués par un atome métallique central entouré d’un certain nombre (2, 3, 4, 5, 6) de molécules ou d’ions qui lui sont attachés par des liaisons chimiques, l’édifice pouvant être au total chargé ou neutre.Ces édifices ont une géométrie simple (fig. 2): tétraèdre, pyramide, octaèdre, carré, pour 4, 5, 6 ou 8 ligands , si l’on appelle ainsi les molécules ou ions liés à l’atome central. Les ligandes sont très variées: ions négatifs tels que Cl-, OH-, C-; molécules telles que H2O, NH3; mais les plus intéressantes pour la catalyse sont par exemple l’oxyde de carbone CO, l’éthylène C2H4, l’acétylène C2H2, l’hydrogène H, les radicaux alcoyles R.En effet, ces corps prennent, dans le complexe, une réactivité particulière: ils sont activés, au sens défini plus haut, et l’atome métallique central constitue un centre actif pour la catalyse.Leur introduction dans le complexe, qui est l’acte fondamental de l’activation, peut se faire par échange de ligandes (substitution), ou par augmentation du nombre de ligandes (addition), processus souvent réversibles. Ainsi, l’activation de la molécule d’hydrogène H2, à froid, est obtenue avec certains complexes de l’argent: [Ag(NR3)2]+ ou du cobalt: [Co(CN)5]3-, en solution aqueuse, avec dissociation de H2 en 2 H, réaction qui ne se produit, en phase gazeuse, qu’à des températures supérieures à 2 000 0C.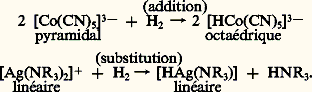 L’oxyde de carbone CO forme volontiers des complexes nommés métaux carbonyles, par attaque directe du métal ou de ses ions:
L’oxyde de carbone CO forme volontiers des complexes nommés métaux carbonyles, par attaque directe du métal ou de ses ions: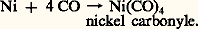 Les métaux carbonyles sont volatils, solubles dans les solvants organiques, et particulièrement aptes à échanger leur CO contre d’autres ligandes. C’est ainsi que l’on peut introduire dans ces complexes des alcènes et des alcynes (fig. 3):
Les métaux carbonyles sont volatils, solubles dans les solvants organiques, et particulièrement aptes à échanger leur CO contre d’autres ligandes. C’est ainsi que l’on peut introduire dans ces complexes des alcènes et des alcynes (fig. 3): Le premier complexe de l’éthylène, trouvé par Zeise (1827), fut obtenu par échange de ligande entre l’éthylène et une solution aqueuse d’acide chloroplatinique (fig. 4):
Le premier complexe de l’éthylène, trouvé par Zeise (1827), fut obtenu par échange de ligande entre l’éthylène et une solution aqueuse d’acide chloroplatinique (fig. 4):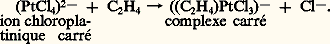 Les molécules ainsi activées par entrée dans le complexe sont particulièrement aptes à réagir, notamment avec les autres ligandes du même complexe.Par exemple, si un complexe est capable à la fois d’activer l’hydrogène et les alcènes, il constituera fort probablement un catalyseur d’hydrogénation. C’est le cas des complexes cyanés du cobalt déjà mentionnés, qui hydrogènent le butadiène en butène:
Les molécules ainsi activées par entrée dans le complexe sont particulièrement aptes à réagir, notamment avec les autres ligandes du même complexe.Par exemple, si un complexe est capable à la fois d’activer l’hydrogène et les alcènes, il constituera fort probablement un catalyseur d’hydrogénation. C’est le cas des complexes cyanés du cobalt déjà mentionnés, qui hydrogènent le butadiène en butène: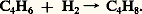 Si un complexe est capable d’activer à la fois l’hydrogène, l’oxyde de carbone et les alcènes, il sera un catalyseur de la réaction bien connue de synthèse des aldéhydes:
Si un complexe est capable d’activer à la fois l’hydrogène, l’oxyde de carbone et les alcènes, il sera un catalyseur de la réaction bien connue de synthèse des aldéhydes: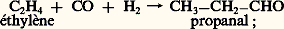 Enfin, un complexe capable d’intégrer plusieurs molécules d’oléfine ou d’acétylène constituera un catalyseur de polymérisation de ces hydrocarbures:
Enfin, un complexe capable d’intégrer plusieurs molécules d’oléfine ou d’acétylène constituera un catalyseur de polymérisation de ces hydrocarbures: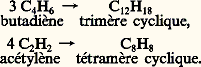 Ces deux corps sont obtenus à l’aide de complexes du nickel, à des températures inférieures à 100 0C. On aura des di-, tri-, tétra-, ... -mères selon que 2, 3, 4, ... molécules de monomère pourront trouver place dans ce complexe.La sélectivité de ces catalyseurs est souvent très grande; c’est ainsi que l’on sait actuellement produire à volonté, à partir du butadiène, ou de l’isoprène, les hauts polymères linéaires à longue chaîne à structure régulière que l’on trouve dans les produits naturels (caoutchouc, gutta-percha, etc.). Elle est d’ailleurs ajustable par un choix judicieux des ligandes inertes du complexe, c’est-à-dire de celles qui ne prennent pas part à la réaction, mais orientent par leur seule géométrie la position des ligandes réactives. Il y a là un champ d’investigation immense ouvert aux chercheurs. La spécificité des processus naturels catalysés par les enzymes dans les organismes vivants a probablement la même origine; d’ailleurs, les centres actifs des enzymes sont aussi des atomes de métaux lourds (Fe, Co, Ni, Cu, Mn) fertiles en complexes. L’industrie des fermentations fait un usage constant de ces catalyseurs naturels, dont la nature est encore mal connue. Mais l’image de la clef et de la serrure, qui est souvent utilisée pour décrire l’ajustement du corps catalysé et du catalyseur, trouve dans ce qui précède une justification scientifique remarquable.4. Catalyse hétérogèneL’action d’un solide qui reste en apparence inaltéré sur la vitesse des réactions chimiques dans un gaz ou un liquide à son contact est restée longtemps inexplicable et hétérogène. C’est pourquoi la catalyse hétérogène, ou catalyse de contact, a été pendant longtemps traitée indépendamment de la catalyse homogène. L’ignorance de la nature physicochimique exacte d’un solide et de sa structure ajoutait à la difficulté; le catalyseur solide ressemblait assez à la «pierre philosophale», qu’on pensait capable de transmuter en or plusieurs millions de fois son poids de vil métal. Les applications industrielles de la catalyse par les solides ont été développées, bien avant que leurs études scientifiques aient abouti à des résultats positifs, et ce fait n’a pas peu contribué à prolonger jusqu’à nos jours cette attitude d’esprit peu rationnelle.Cependant, dès le début du siècle, les chimistes et les physiciens s’accordaient à reconnaître la nature chimique de la catalyse de contact.Sabatier écrivait en 1911: «La catalyse nous semble donc en général résulter de phénomènes purement chimiques accomplis grâce à la présence du catalyseur qui donne, avec l’un des éléments du système primitif, une combinaison temporaire instable dont la destruction ou la réaction rapide sur les autres éléments déterminent la transformation du système, le catalyseur étant régénéré semblable à lui-même, capable de poursuivre indéfiniment le même effet.» Ce point de vue était opposé à l’explication dite «physique» de la catalyse par l’adsorption dans les pores du solide, qui provoquerait une concentration locale élevée de réactifs. Nous savons maintenant que les phénomènes d’adsorption sont complexes, et qu’il faut distinguer la rétention des molécules à la surface de tous les solides par des forces de Van der Waals analogues aux forces qui s’exercent entre les molécules d’un gaz ou d’un liquide – forces faibles et peu spécifiques – de l’adsorption chimique qui est très énergique et très spécifique, car elle crée des liaisons chimiques entre la molécule et les atomes superficiels du solide. Cette chimisorption crée les combinaisons temporaires instables dont parle Sabatier, et active la molécule, qui reste dans son état normal lors de l’adsorption purement physique. Langmuir écrivait vers la même époque:«Les atomes formant la surface d’un solide sont liés à ceux des couches inférieures par des forces semblables à celles qui s’exercent entre les atomes de la partie interne. Depuis les travaux de Bragg sur la structure cristalline corroborés par de nombreuses autres considérations nous savons que ces forces sont de nature chimique. Dans la couche superficielle, à cause de l’asymétrie des conditions, l’arrangement des atomes doit être toujours légèrement différent de celui de l’intérieur. Ces atomes sont chimiquement insaturés et donc environnés d’un intense champ de forces.»«D’autres considérations m’amenèrent à penser que, lorsque les molécules de gaz entrent en collision avec n’importe quelle surface solide ou liquide, elles ne rebondissent pas en général de façon élastique mais se condensent à la surface, étant retenues par le champ de forces des atomes de cette surface. Ces molécules peuvent ensuite s’évaporer. Le temps qui s’écoule entre la condensation d’une molécule et son évaporation dépend de l’intensité des forces superficielles. L’adsorption est le résultat direct de cet intervalle de temps. Si les forces superficielles sont relativement intenses, l’évaporation aura lieu à une vitesse négligeable, si bien que la surface du solide sera complètement recouverte d’une couche de molécules. Dans le cas d’une adsorption vraie, cette couche n’aura en général pas plus d’une molécule d’épaisseur, car aussitôt que la surface est recouverte d’une monocouche, les forces superficielles sont chimiquement saturées» (1915-1918). La notion de centre actif superficiel se trouve aussi dans les écrits de Langmuir.Les faces planes d’un cristal doivent être constituées d’atomes formant un réseau plan régulier. La surface des cristaux ressemble donc dans une certaine mesure à un damier.Quand les molécules de gaz sont adsorbées par une telle surface, elles adoptent des positions définies par rapport au réseau de la surface et tendent ainsi à former par-dessus un nouveau réseau.Par conséquent, une surface unitaire de n’importe quelle surface cristalline a un nombre défini «d’espaces élémentaires» capable chacun de fixer une molécule ou un atome adsorbé. En général, ces sites ne seront pas exactement identiques.C’est introduire l’idée qu’une même surface porte des distributions de centres actifs de propriétés différentes. La différence essentielle entre catalyseurs homogènes et hétérogènes est parfaitement claire: dans le premier cas, les centres actifs sont libres; dans le deuxième, ils sont liés à la surface d’un solide. Cinquante années de travaux sur la catalyse n’ont fait que préciser ces idées et confirmer que, dans beaucoup de cas, les centres actifs superficiels ont des analogues libres. C’est ainsi que l’on trouve des centres acides ou basiques à la surface d’oxydes simples ou complexes tels que l’alumine, les silicates d’alumine, les phosphates, etc., et des centres actifs activant l’hydrogène à la surface des métaux de transition ou de leurs composés binaires solides: oxydes, sulfures.La nature de ces centres est parfaitement déterminée par celle des molécules qu’ils activent, et peut être rattachée à la structure maintenant connue de la plupart des solides.5. Particularités des catalyseurs solidesLa catalyse étant un phénomène de surface, l’activité d’un solide sera en général d’autant plus grande que le rapport surface/masse, ou aire spécifique (m2/g) sera plus élevé, c’est-à-dire que le solide sera plus poreux et divisé. L’art de la préparation des catalyseurs contient de nombreuses recettes à cette fin. En général, le solide sera d’autant plus divisé qu’il sera élaboré à une température plus basse: on pourra opérer par voie humide en formant un précipité à partir d’une solution, ou par voie sèche en attaquant un solide par un gaz. Les moyens mécaniques de broyage sont assez peu utilisés. À titre d’exemple, l’aire spécifique d’un catalyseur peut aller de quelques m2/g à plusieurs centaines de m2/g:
Ces deux corps sont obtenus à l’aide de complexes du nickel, à des températures inférieures à 100 0C. On aura des di-, tri-, tétra-, ... -mères selon que 2, 3, 4, ... molécules de monomère pourront trouver place dans ce complexe.La sélectivité de ces catalyseurs est souvent très grande; c’est ainsi que l’on sait actuellement produire à volonté, à partir du butadiène, ou de l’isoprène, les hauts polymères linéaires à longue chaîne à structure régulière que l’on trouve dans les produits naturels (caoutchouc, gutta-percha, etc.). Elle est d’ailleurs ajustable par un choix judicieux des ligandes inertes du complexe, c’est-à-dire de celles qui ne prennent pas part à la réaction, mais orientent par leur seule géométrie la position des ligandes réactives. Il y a là un champ d’investigation immense ouvert aux chercheurs. La spécificité des processus naturels catalysés par les enzymes dans les organismes vivants a probablement la même origine; d’ailleurs, les centres actifs des enzymes sont aussi des atomes de métaux lourds (Fe, Co, Ni, Cu, Mn) fertiles en complexes. L’industrie des fermentations fait un usage constant de ces catalyseurs naturels, dont la nature est encore mal connue. Mais l’image de la clef et de la serrure, qui est souvent utilisée pour décrire l’ajustement du corps catalysé et du catalyseur, trouve dans ce qui précède une justification scientifique remarquable.4. Catalyse hétérogèneL’action d’un solide qui reste en apparence inaltéré sur la vitesse des réactions chimiques dans un gaz ou un liquide à son contact est restée longtemps inexplicable et hétérogène. C’est pourquoi la catalyse hétérogène, ou catalyse de contact, a été pendant longtemps traitée indépendamment de la catalyse homogène. L’ignorance de la nature physicochimique exacte d’un solide et de sa structure ajoutait à la difficulté; le catalyseur solide ressemblait assez à la «pierre philosophale», qu’on pensait capable de transmuter en or plusieurs millions de fois son poids de vil métal. Les applications industrielles de la catalyse par les solides ont été développées, bien avant que leurs études scientifiques aient abouti à des résultats positifs, et ce fait n’a pas peu contribué à prolonger jusqu’à nos jours cette attitude d’esprit peu rationnelle.Cependant, dès le début du siècle, les chimistes et les physiciens s’accordaient à reconnaître la nature chimique de la catalyse de contact.Sabatier écrivait en 1911: «La catalyse nous semble donc en général résulter de phénomènes purement chimiques accomplis grâce à la présence du catalyseur qui donne, avec l’un des éléments du système primitif, une combinaison temporaire instable dont la destruction ou la réaction rapide sur les autres éléments déterminent la transformation du système, le catalyseur étant régénéré semblable à lui-même, capable de poursuivre indéfiniment le même effet.» Ce point de vue était opposé à l’explication dite «physique» de la catalyse par l’adsorption dans les pores du solide, qui provoquerait une concentration locale élevée de réactifs. Nous savons maintenant que les phénomènes d’adsorption sont complexes, et qu’il faut distinguer la rétention des molécules à la surface de tous les solides par des forces de Van der Waals analogues aux forces qui s’exercent entre les molécules d’un gaz ou d’un liquide – forces faibles et peu spécifiques – de l’adsorption chimique qui est très énergique et très spécifique, car elle crée des liaisons chimiques entre la molécule et les atomes superficiels du solide. Cette chimisorption crée les combinaisons temporaires instables dont parle Sabatier, et active la molécule, qui reste dans son état normal lors de l’adsorption purement physique. Langmuir écrivait vers la même époque:«Les atomes formant la surface d’un solide sont liés à ceux des couches inférieures par des forces semblables à celles qui s’exercent entre les atomes de la partie interne. Depuis les travaux de Bragg sur la structure cristalline corroborés par de nombreuses autres considérations nous savons que ces forces sont de nature chimique. Dans la couche superficielle, à cause de l’asymétrie des conditions, l’arrangement des atomes doit être toujours légèrement différent de celui de l’intérieur. Ces atomes sont chimiquement insaturés et donc environnés d’un intense champ de forces.»«D’autres considérations m’amenèrent à penser que, lorsque les molécules de gaz entrent en collision avec n’importe quelle surface solide ou liquide, elles ne rebondissent pas en général de façon élastique mais se condensent à la surface, étant retenues par le champ de forces des atomes de cette surface. Ces molécules peuvent ensuite s’évaporer. Le temps qui s’écoule entre la condensation d’une molécule et son évaporation dépend de l’intensité des forces superficielles. L’adsorption est le résultat direct de cet intervalle de temps. Si les forces superficielles sont relativement intenses, l’évaporation aura lieu à une vitesse négligeable, si bien que la surface du solide sera complètement recouverte d’une couche de molécules. Dans le cas d’une adsorption vraie, cette couche n’aura en général pas plus d’une molécule d’épaisseur, car aussitôt que la surface est recouverte d’une monocouche, les forces superficielles sont chimiquement saturées» (1915-1918). La notion de centre actif superficiel se trouve aussi dans les écrits de Langmuir.Les faces planes d’un cristal doivent être constituées d’atomes formant un réseau plan régulier. La surface des cristaux ressemble donc dans une certaine mesure à un damier.Quand les molécules de gaz sont adsorbées par une telle surface, elles adoptent des positions définies par rapport au réseau de la surface et tendent ainsi à former par-dessus un nouveau réseau.Par conséquent, une surface unitaire de n’importe quelle surface cristalline a un nombre défini «d’espaces élémentaires» capable chacun de fixer une molécule ou un atome adsorbé. En général, ces sites ne seront pas exactement identiques.C’est introduire l’idée qu’une même surface porte des distributions de centres actifs de propriétés différentes. La différence essentielle entre catalyseurs homogènes et hétérogènes est parfaitement claire: dans le premier cas, les centres actifs sont libres; dans le deuxième, ils sont liés à la surface d’un solide. Cinquante années de travaux sur la catalyse n’ont fait que préciser ces idées et confirmer que, dans beaucoup de cas, les centres actifs superficiels ont des analogues libres. C’est ainsi que l’on trouve des centres acides ou basiques à la surface d’oxydes simples ou complexes tels que l’alumine, les silicates d’alumine, les phosphates, etc., et des centres actifs activant l’hydrogène à la surface des métaux de transition ou de leurs composés binaires solides: oxydes, sulfures.La nature de ces centres est parfaitement déterminée par celle des molécules qu’ils activent, et peut être rattachée à la structure maintenant connue de la plupart des solides.5. Particularités des catalyseurs solidesLa catalyse étant un phénomène de surface, l’activité d’un solide sera en général d’autant plus grande que le rapport surface/masse, ou aire spécifique (m2/g) sera plus élevé, c’est-à-dire que le solide sera plus poreux et divisé. L’art de la préparation des catalyseurs contient de nombreuses recettes à cette fin. En général, le solide sera d’autant plus divisé qu’il sera élaboré à une température plus basse: on pourra opérer par voie humide en formant un précipité à partir d’une solution, ou par voie sèche en attaquant un solide par un gaz. Les moyens mécaniques de broyage sont assez peu utilisés. À titre d’exemple, l’aire spécifique d’un catalyseur peut aller de quelques m2/g à plusieurs centaines de m2/g: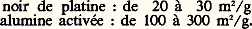 Pour les solides divisés non poreux, l’aire spécifique est en relation simple avec la dimension moyenne des grains. Ainsi, pour le platine de masse volumique 21,45 g/cm3, en supposant les grains cubiques, l’arête moyenne des cubes serait de 10,0 à 14,5 nm dans l’exemple cité, soit 35 à 50 atomes. On peut diminuer fortement ces dimensions en dispersant le métal sur un support poreux comme une alumine, et obtenir ainsi des aires spécifiques de 50 à 100 m2/g de platine, soit des tailles de 2,5 à 5,5 nm. Pour les solides poreux, l’aire spécifique est liée à la finesse des pores, qui peuvent descendre à quelques nanomètres. Une grande aire spécifique est d’autant moins nécessaire que le catalyseur est plus actif, et il est possible, par exemple, dans certaines réactions, d’utiliser le platine en fils (toiles).Mais l’aire spécifique donne la quantité de surface active et non sa qualité, c’est-à-dire la nature et la densité des centres actifs. Cette qualité dépend énormément de la méthode d’élaboration du catalyseur. Par définition, les centres actifs sont les atomes ou groupes d’atomes de la surface les plus propres à former des combinaisons chimiques, et, en l’absence de précautions spéciales, ils se trouveront tous saturés lors de la préparation du catalyseur.En effet, le solide trouvera toujours, soit dans la solution mère, soit dans l’atmosphère qui l’entoure, des molécules disposées à se combiner avec les atomes de sa surface. Par exemple, un solide tel que le nickel divisé adsorbera avec beaucoup de ténacité l’oxygène de l’air, et les centres acides d’une alumine préparée par voie aqueuse seront neutralisés par les ions basiques.Le traitement préliminaire activant qui est, par exemple, une réduction par l’hydrogène, ou une déshydratation par chauffage, sera donc essentiel.Il est clair que la simple spécification de la nature chimique d’un catalyseur est très insuffisante pour le caractériser, car ses propriétés finales dépendront, comme on l’a vu, de tous les processus employés pour sa fabrication.La durée de vie d’un catalyseur solide est limitée par celle de ses centres actifs. Leur disparition (désactivation) peut avoir de nombreuses causes. Parmi les plus fréquentes, on citera la chute de l’aire spécifique par coalescence des grains, ou frittage , provoqué par une température trop élevée: c’est un processus physique irréversible, qui est dû à l’instabilité thermodynamique des petits grains par rapport aux gros (énergie superficielle).L’empoisonnement du catalyseur se produit lorsqu’on introduit accidentellement dans le milieu un corps, dit poison, dont les molécules sont très fortement adsorbées par les centres actifs, qui se trouvent ainsi bloqués. Les composés du soufre (hydrogène sulfuré, sulfure de carbone, thiols) sont ainsi des poisons violents pour les métaux (Ni, Pt).L’encrassement du catalyseur est observé lorsqu’une réaction secondaire forme à sa surface des produits indésorbables de grande masse moléculaire. Ainsi, dans de nombreux traitements catalytiques des hydrocarbures, des produits lourds, mal définis sous le nom de coke, bloquent lentement les centres actifs. L’empoisonnement et l’encrassement sont dits réversibles lorsque par un traitement approprié (autre que la destruction totale du catalyseur en ses éléments) on peut débloquer les centres actifs mis hors d’usage. Ainsi, un dépôt de coke peut être enlevé par combustion ménagée à l’oxygène dilué, suivi éventuellement d’une réduction, si on a un métal dans le catalyseur.Composition et aspect des catalyseurs solidesLes catalyseurs utilisés en pratique sont fréquemment des mélanges où l’on peut distinguer le composant actif , le support et les promoteurs .Ces derniers sont des composants par eux-mêmes inactifs, mais capables, par divers moyens, d’améliorer la stabilité, la quantité ou la qualité des centres actifs.Certains empêchent le frittage de la phase active et conservent une aire spécifique élevée par simple insertion entre les grains de cette phase: ainsi, un faible pourcentage d’oxyde de chrome ou d’aluminium prolonge considérablement la vie du nickel divisé. D’autres se combinent ou se dissolvent dans la phase active et modifient la nature même des centres actifs, par un effet qui n’est pas sans analogie avec celui du changement des ligandes inertes dans les catalyseurs solubles (cf. Catalyseurs complexes ). Il y a promotion lorsque cette addition augmente l’activité ou la sélectivité du catalyseur, et l’on observe toujours un maximum de ces grandeurs quand la dose de promoteur augmente (fig. 5). Lorsque la quantité optimale de promoteur est élevée, on parle de catalyseur mixte: ainsi, les oxydes mixtes de vanadium et de molybdène ont une sélectivité maximale pour l’oxydation du benzène en anhydride maléique (base de matières plastiques du type polyester) pour environ V25/Mo3 = 3.Les catalyseurs se présentent en poudres, grains, sphérules, ou pastilles de dimensions variant de quelques centaines de microns à plusieurs centimètres. Les plus gros sont employés en lit fixe , couches d’épaisseur variable, traversées par le fluide, liquide ou gaz, tandis que les plus fins sont mis en suspension dans ce fluide, dont il faut les séparer (décantation, filtration...). Dans tous les cas, les produits de l’opération sont plus faciles à séparer du catalyseur que dans la catalyse homogène.En pratique, la durée de vie du catalyseur est très importante, car elle règle l’incidence du prix du catalyseur sur les prix de revient de la fabrication. Il résulte de ce qui précède que cette durée de vie augmentera si on abaisse la température de travail et si on augmente la pureté des corps introduits.La consommation de catalyseur par tonne de produit traité (pertes ou désactivation irréversible) peut varier de 1 kg (craquage catalytique) à 1 g environ (synthèse de l’ammoniac).Cinétique de la catalyse hétérogèneLa cinétique des réactions catalysées est dominée par la vitesse de réaction des centres actifs avec les corps de départ ou la vitesse de libération de ces centres par désorption des produits; ces deux vitesses étant égales lorsque s’est établi le régime stationnaire, l’étape la plus lente gouverne la vitesse globale. Cette compétition pour les centres actifs se traduit souvent par un effet de ralentissement de la réaction par ses propres produits, appelé auto-inhibition .Ainsi, la synthèse de l’ammoniac est inhibée par l’ammoniac, et sa décomposition est inhibée par l’hydrogène:
Pour les solides divisés non poreux, l’aire spécifique est en relation simple avec la dimension moyenne des grains. Ainsi, pour le platine de masse volumique 21,45 g/cm3, en supposant les grains cubiques, l’arête moyenne des cubes serait de 10,0 à 14,5 nm dans l’exemple cité, soit 35 à 50 atomes. On peut diminuer fortement ces dimensions en dispersant le métal sur un support poreux comme une alumine, et obtenir ainsi des aires spécifiques de 50 à 100 m2/g de platine, soit des tailles de 2,5 à 5,5 nm. Pour les solides poreux, l’aire spécifique est liée à la finesse des pores, qui peuvent descendre à quelques nanomètres. Une grande aire spécifique est d’autant moins nécessaire que le catalyseur est plus actif, et il est possible, par exemple, dans certaines réactions, d’utiliser le platine en fils (toiles).Mais l’aire spécifique donne la quantité de surface active et non sa qualité, c’est-à-dire la nature et la densité des centres actifs. Cette qualité dépend énormément de la méthode d’élaboration du catalyseur. Par définition, les centres actifs sont les atomes ou groupes d’atomes de la surface les plus propres à former des combinaisons chimiques, et, en l’absence de précautions spéciales, ils se trouveront tous saturés lors de la préparation du catalyseur.En effet, le solide trouvera toujours, soit dans la solution mère, soit dans l’atmosphère qui l’entoure, des molécules disposées à se combiner avec les atomes de sa surface. Par exemple, un solide tel que le nickel divisé adsorbera avec beaucoup de ténacité l’oxygène de l’air, et les centres acides d’une alumine préparée par voie aqueuse seront neutralisés par les ions basiques.Le traitement préliminaire activant qui est, par exemple, une réduction par l’hydrogène, ou une déshydratation par chauffage, sera donc essentiel.Il est clair que la simple spécification de la nature chimique d’un catalyseur est très insuffisante pour le caractériser, car ses propriétés finales dépendront, comme on l’a vu, de tous les processus employés pour sa fabrication.La durée de vie d’un catalyseur solide est limitée par celle de ses centres actifs. Leur disparition (désactivation) peut avoir de nombreuses causes. Parmi les plus fréquentes, on citera la chute de l’aire spécifique par coalescence des grains, ou frittage , provoqué par une température trop élevée: c’est un processus physique irréversible, qui est dû à l’instabilité thermodynamique des petits grains par rapport aux gros (énergie superficielle).L’empoisonnement du catalyseur se produit lorsqu’on introduit accidentellement dans le milieu un corps, dit poison, dont les molécules sont très fortement adsorbées par les centres actifs, qui se trouvent ainsi bloqués. Les composés du soufre (hydrogène sulfuré, sulfure de carbone, thiols) sont ainsi des poisons violents pour les métaux (Ni, Pt).L’encrassement du catalyseur est observé lorsqu’une réaction secondaire forme à sa surface des produits indésorbables de grande masse moléculaire. Ainsi, dans de nombreux traitements catalytiques des hydrocarbures, des produits lourds, mal définis sous le nom de coke, bloquent lentement les centres actifs. L’empoisonnement et l’encrassement sont dits réversibles lorsque par un traitement approprié (autre que la destruction totale du catalyseur en ses éléments) on peut débloquer les centres actifs mis hors d’usage. Ainsi, un dépôt de coke peut être enlevé par combustion ménagée à l’oxygène dilué, suivi éventuellement d’une réduction, si on a un métal dans le catalyseur.Composition et aspect des catalyseurs solidesLes catalyseurs utilisés en pratique sont fréquemment des mélanges où l’on peut distinguer le composant actif , le support et les promoteurs .Ces derniers sont des composants par eux-mêmes inactifs, mais capables, par divers moyens, d’améliorer la stabilité, la quantité ou la qualité des centres actifs.Certains empêchent le frittage de la phase active et conservent une aire spécifique élevée par simple insertion entre les grains de cette phase: ainsi, un faible pourcentage d’oxyde de chrome ou d’aluminium prolonge considérablement la vie du nickel divisé. D’autres se combinent ou se dissolvent dans la phase active et modifient la nature même des centres actifs, par un effet qui n’est pas sans analogie avec celui du changement des ligandes inertes dans les catalyseurs solubles (cf. Catalyseurs complexes ). Il y a promotion lorsque cette addition augmente l’activité ou la sélectivité du catalyseur, et l’on observe toujours un maximum de ces grandeurs quand la dose de promoteur augmente (fig. 5). Lorsque la quantité optimale de promoteur est élevée, on parle de catalyseur mixte: ainsi, les oxydes mixtes de vanadium et de molybdène ont une sélectivité maximale pour l’oxydation du benzène en anhydride maléique (base de matières plastiques du type polyester) pour environ V25/Mo3 = 3.Les catalyseurs se présentent en poudres, grains, sphérules, ou pastilles de dimensions variant de quelques centaines de microns à plusieurs centimètres. Les plus gros sont employés en lit fixe , couches d’épaisseur variable, traversées par le fluide, liquide ou gaz, tandis que les plus fins sont mis en suspension dans ce fluide, dont il faut les séparer (décantation, filtration...). Dans tous les cas, les produits de l’opération sont plus faciles à séparer du catalyseur que dans la catalyse homogène.En pratique, la durée de vie du catalyseur est très importante, car elle règle l’incidence du prix du catalyseur sur les prix de revient de la fabrication. Il résulte de ce qui précède que cette durée de vie augmentera si on abaisse la température de travail et si on augmente la pureté des corps introduits.La consommation de catalyseur par tonne de produit traité (pertes ou désactivation irréversible) peut varier de 1 kg (craquage catalytique) à 1 g environ (synthèse de l’ammoniac).Cinétique de la catalyse hétérogèneLa cinétique des réactions catalysées est dominée par la vitesse de réaction des centres actifs avec les corps de départ ou la vitesse de libération de ces centres par désorption des produits; ces deux vitesses étant égales lorsque s’est établi le régime stationnaire, l’étape la plus lente gouverne la vitesse globale. Cette compétition pour les centres actifs se traduit souvent par un effet de ralentissement de la réaction par ses propres produits, appelé auto-inhibition .Ainsi, la synthèse de l’ammoniac est inhibée par l’ammoniac, et sa décomposition est inhibée par l’hydrogène:
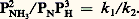 La phase fluide n’intervient que comme un réservoir de réactifs et de produits, qui sont respectivement amenés vers la surface, ou en sens inverse, par la diffusion des molécules. Si la réaction de surface est très rapide, la vitesse globale sera limitée par celle de ces transferts et, à la limite, deviendra indépendante de l’activié du catalyseur. Cette limitation diffusionnelle joue un grand rôle lorsque les centres actifs sont sur les parois internes de pores très fins (micropores de quelques dizaines d’angströms de diamètre), à travers lesquels la diffusion est lente. La partie centrale d’un grain de catalyseur microporeux risque ainsi de ne pas être du tout utilisée, les molécules des réactifs étant complètement transformées avant de les atteindre. Une aire spécifique trop élevée n’est pas toujours avantageuse, notamment pour les catalyseurs très actifs. L’activité sera mesurée ici en moles converties par unité de temps et unité de surface active. Quand la chose est possible, la densité superficielle de centres actifs est une indication très précieuse. Elle est d’environ 1015 par cm2 sur les métaux (Ni, Pt, Fe) et de 1012 par cm2 sur les oxydes métalliques. On peut l’estimer par des mesures d’adsorption chimique.Les grandes familles de catalyseurs solidesLes catalyseurs solides sont en général des métaux ou des composés binaires ou ternaires de ces métaux avec des éléments non métalliques et, de façon plus générale, des solides à point de fusion élevé, indiquant de fortes liaisons chimiques entre leurs atomes ou ions. Comme le note Langmuir (cf. chap. 4), le clivage de tels solides crée une surface hautement insaturée au point de vue chimique, et, partant, des centres actifs. On trouve assez souvent que ces centres actifs ont des propriétés semblables à celles de certains catalyseurs homogènes, et accélèrent les mêmes réactions.Une première classe est constituée par les solides qui activent l’hydrogène, l’oxyde de carbone, l’éthylène, l’oxygène: ce sont les métaux de transition et leurs oxydes, sulfures... qui donnent ainsi les complexes solubles ayant des propriétés catalytiques similaires. Leurs centres actifs sont construits autour de l’atome de transition, chargé ou non, qui permet la formation d’un grand nombre de liaisons (coordinance variable). Que cet atome soit au centre d’une molécule complexe ou dans un solide, sa configuration électronique reste la même; aussi, une molécule comme CO liée dans un métal-carbonyle ou à un atome de surface du métal cristallisé, ou à un ion superficiel de son oxyde se trouve-t-elle dans un état actif (fig. 6). Les différences constatées peuvent s’expliquer par l’influence des ligandes inertes ou des atomes qui les remplacent dans les solides.On trouve dans cette classe les catalyseurs d’hydrogénation, comme le nickel, le platine, le fer, le cobalt, l’oxyde de nickel, l’oxyde de chrome, etc., qui sont aussi des catalyseurs de déshydrogénation et d’hydrogénolyse, car ils activent notamment les liaisons H-H, C-H, O-H, N-H en séparant le H.Les catalyseurs d’oxydation, qui activent l’oxygène 2 tels que le platine, les oxydes de manganèse, cobalt, cuivre, vanadium, molybdène, tungstène appartiennent également à cette classe. Leurs centres actifs se combinent aussi volontiers avec les corps organiques insaturés: éthylène, benzène et dérivés, et permettent leur hydrogénation et leur oxydation.Une deuxième classe comprend les oxydes acides ou basiques dont l’activité catalytique est semblable à celle des acides ou bases solubles, et qui activent les molécules ionisables, par exemple par addition ou soustraction d’un proton H+. On peut d’ailleurs titrer l’acidité des catalyseurs solides par les mêmes indicateurs colorés d’emploi courant en solution aqueuse, et mesurer leur force. Les centres acides à la surface des catalyseurs mixtes silice-alumine sont aussi forts que l’acide sulfurique à 90 p. 100: cela s’explique par la présence de groupes fonctionnels semblables aux anions des oxyacides solubles. L’analogie entre Al4- (sur les silices-alumines) et Cl4- (solutions d’acide perchlorique) mérite d’être soulignée (fig. 7).Quant aux centres basiques, ils peuvent être identifiés aux anions de surface, par exemple 2-, dans les oxydes, lorsque ces anions sont incomplètement saturés.Cette classe inclut un grand nombre de silicates, phosphates, borates, les alumines, la thorine. Le groupe des silicates d’aluminium synthétiques ou naturels est particulièrement riche; leurs centres actifs acides sont les groupes Al4- H+ dus à la dispersion de l’aluminium dans un réseau de silice. Les argiles naturelles activées, les tamis moléculaires naturels ou artificiels en font partie.L’activation de l’eau et des oléfines permet de réaliser les réactions d’hydratation, de déshydratation, d’alcoylation, de craquage, de polymérisation, d’isomérisation des corps organiques.En conclusion, il faut souligner l’importance pratique de la catalyse dans l’industrie chimique et donc dans la vie économique moderne. La découverte d’un catalyseur nouveau peut bouleverser la structure de toute une industrie, voire même le cours de notre civilisation. À ce titre, le catalyseur est vraiment la version moderne de la pierre philosophale.
La phase fluide n’intervient que comme un réservoir de réactifs et de produits, qui sont respectivement amenés vers la surface, ou en sens inverse, par la diffusion des molécules. Si la réaction de surface est très rapide, la vitesse globale sera limitée par celle de ces transferts et, à la limite, deviendra indépendante de l’activié du catalyseur. Cette limitation diffusionnelle joue un grand rôle lorsque les centres actifs sont sur les parois internes de pores très fins (micropores de quelques dizaines d’angströms de diamètre), à travers lesquels la diffusion est lente. La partie centrale d’un grain de catalyseur microporeux risque ainsi de ne pas être du tout utilisée, les molécules des réactifs étant complètement transformées avant de les atteindre. Une aire spécifique trop élevée n’est pas toujours avantageuse, notamment pour les catalyseurs très actifs. L’activité sera mesurée ici en moles converties par unité de temps et unité de surface active. Quand la chose est possible, la densité superficielle de centres actifs est une indication très précieuse. Elle est d’environ 1015 par cm2 sur les métaux (Ni, Pt, Fe) et de 1012 par cm2 sur les oxydes métalliques. On peut l’estimer par des mesures d’adsorption chimique.Les grandes familles de catalyseurs solidesLes catalyseurs solides sont en général des métaux ou des composés binaires ou ternaires de ces métaux avec des éléments non métalliques et, de façon plus générale, des solides à point de fusion élevé, indiquant de fortes liaisons chimiques entre leurs atomes ou ions. Comme le note Langmuir (cf. chap. 4), le clivage de tels solides crée une surface hautement insaturée au point de vue chimique, et, partant, des centres actifs. On trouve assez souvent que ces centres actifs ont des propriétés semblables à celles de certains catalyseurs homogènes, et accélèrent les mêmes réactions.Une première classe est constituée par les solides qui activent l’hydrogène, l’oxyde de carbone, l’éthylène, l’oxygène: ce sont les métaux de transition et leurs oxydes, sulfures... qui donnent ainsi les complexes solubles ayant des propriétés catalytiques similaires. Leurs centres actifs sont construits autour de l’atome de transition, chargé ou non, qui permet la formation d’un grand nombre de liaisons (coordinance variable). Que cet atome soit au centre d’une molécule complexe ou dans un solide, sa configuration électronique reste la même; aussi, une molécule comme CO liée dans un métal-carbonyle ou à un atome de surface du métal cristallisé, ou à un ion superficiel de son oxyde se trouve-t-elle dans un état actif (fig. 6). Les différences constatées peuvent s’expliquer par l’influence des ligandes inertes ou des atomes qui les remplacent dans les solides.On trouve dans cette classe les catalyseurs d’hydrogénation, comme le nickel, le platine, le fer, le cobalt, l’oxyde de nickel, l’oxyde de chrome, etc., qui sont aussi des catalyseurs de déshydrogénation et d’hydrogénolyse, car ils activent notamment les liaisons H-H, C-H, O-H, N-H en séparant le H.Les catalyseurs d’oxydation, qui activent l’oxygène 2 tels que le platine, les oxydes de manganèse, cobalt, cuivre, vanadium, molybdène, tungstène appartiennent également à cette classe. Leurs centres actifs se combinent aussi volontiers avec les corps organiques insaturés: éthylène, benzène et dérivés, et permettent leur hydrogénation et leur oxydation.Une deuxième classe comprend les oxydes acides ou basiques dont l’activité catalytique est semblable à celle des acides ou bases solubles, et qui activent les molécules ionisables, par exemple par addition ou soustraction d’un proton H+. On peut d’ailleurs titrer l’acidité des catalyseurs solides par les mêmes indicateurs colorés d’emploi courant en solution aqueuse, et mesurer leur force. Les centres acides à la surface des catalyseurs mixtes silice-alumine sont aussi forts que l’acide sulfurique à 90 p. 100: cela s’explique par la présence de groupes fonctionnels semblables aux anions des oxyacides solubles. L’analogie entre Al4- (sur les silices-alumines) et Cl4- (solutions d’acide perchlorique) mérite d’être soulignée (fig. 7).Quant aux centres basiques, ils peuvent être identifiés aux anions de surface, par exemple 2-, dans les oxydes, lorsque ces anions sont incomplètement saturés.Cette classe inclut un grand nombre de silicates, phosphates, borates, les alumines, la thorine. Le groupe des silicates d’aluminium synthétiques ou naturels est particulièrement riche; leurs centres actifs acides sont les groupes Al4- H+ dus à la dispersion de l’aluminium dans un réseau de silice. Les argiles naturelles activées, les tamis moléculaires naturels ou artificiels en font partie.L’activation de l’eau et des oléfines permet de réaliser les réactions d’hydratation, de déshydratation, d’alcoylation, de craquage, de polymérisation, d’isomérisation des corps organiques.En conclusion, il faut souligner l’importance pratique de la catalyse dans l’industrie chimique et donc dans la vie économique moderne. La découverte d’un catalyseur nouveau peut bouleverser la structure de toute une industrie, voire même le cours de notre civilisation. À ce titre, le catalyseur est vraiment la version moderne de la pierre philosophale.
Encyclopédie Universelle. 2012.